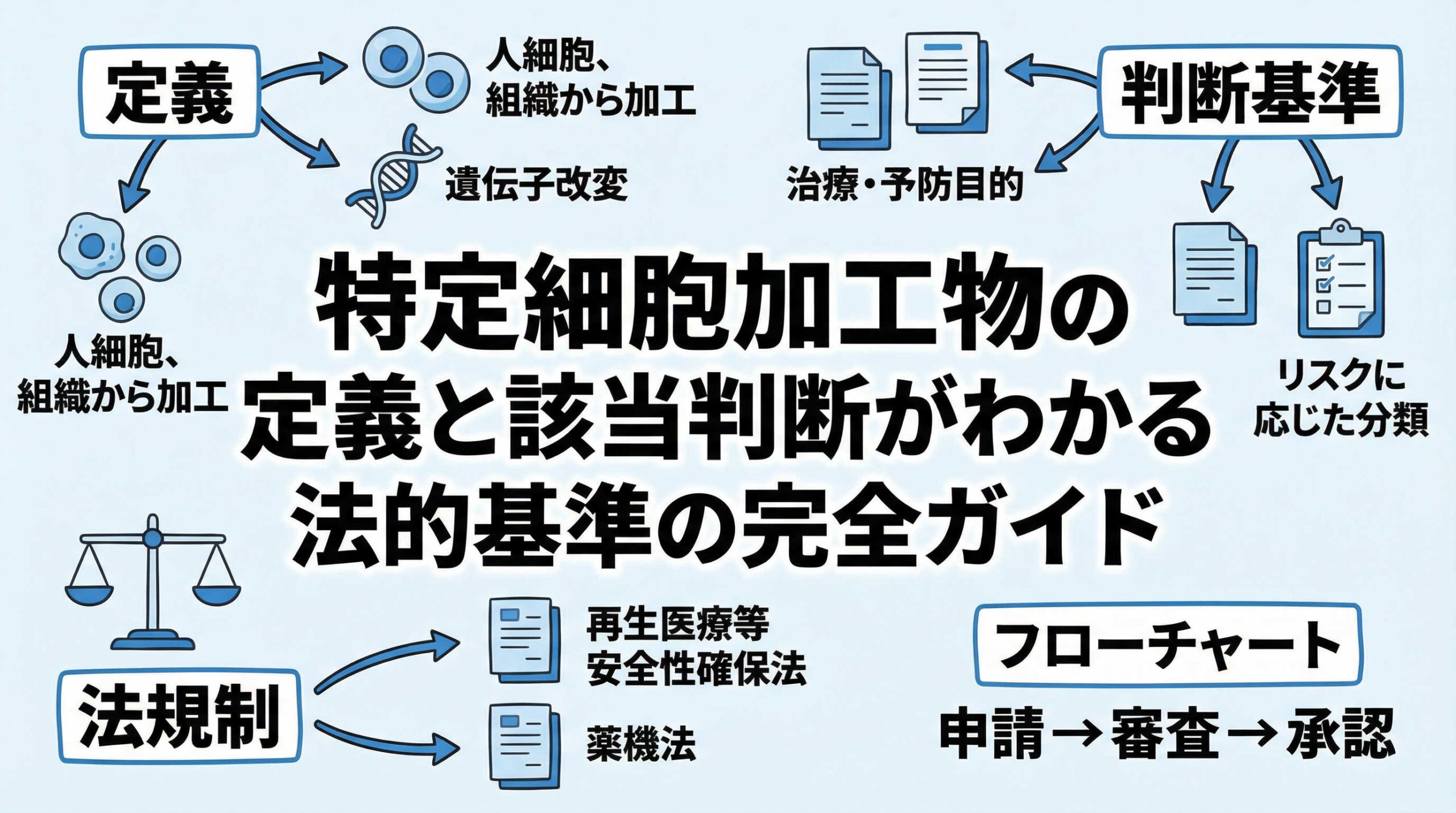
再生医療の現場において、取り扱う細胞が法の規制対象となる「特定細胞加工物」に該当するかどうかの判断は、極めて重要な第一歩です。この定義を正確に理解していなければ、知らず知らずのうちに法的な義務を怠り、コンプライアンス違反のリスクを負うことになりかねません。特に、新規の細胞加工物の取り扱いを検討されている薬事担当者様や医療機関の管理者様にとって、その境界線は時に曖昧で悩ましいものでしょう。
本記事では、再生医療等の安全性の確保等に関する法律(再生医療安全性確保法)に基づき、「特定細胞加工物の定義」を明確に解説いたします。どのような処理が「加工」とみなされ、何が「最小限の操作」として除外されるのか、具体的な判断基準を詳しく紐解いてまいります。自社のケースが該当するかどうかを適切に判定し、安全かつ円滑な事業運営にお役立てください。
特定細胞加工物とは?再生医療安全性確保法に基づく法的定義の結論

再生医療等の安全性の確保等に関する法律(以下、再生医療安全性確保法)において、「特定細胞加工物」がどのように定義されているかを正確に把握することは、再生医療事業の根幹に関わる重要なプロセスです。法的な定義は一見複雑に見えますが、その本質を理解すれば、自社が取り扱う細胞が規制の対象となるか否かを論理的に導き出すことが可能です。ここでは、法第2条第4項に基づく定義と、実務上で特に重要となる「製造」や「調製」の概念について、その核心部分を解説いたします。
再生医療等の安全性の確保等に関する法律(法第2条第4項)による定義
再生医療安全性確保法第2条第4項において、特定細胞加工物は「人又は動物の細胞に培養等の加工を施したもの」であり、かつ「再生医療等に用いるもの」と定義されています。ここで重要なのは、単に細胞を取り出すだけでなく、そこに何らかの人為的な「加工」が加えられているという点です。
具体的には、以下のいずれかに該当するものが対象となります。
- 身体の構造又は機能の再建、修復又は形成を目的とするもの
- 疾病の治療又は予防を目的とするもの
ただし、すべての加工細胞が含まれるわけではなく、医薬品医療機器等法の規定により承認された再生医療等製品などは除かれる場合があるため、法の適用範囲を慎重に確認する必要があります。
「製造」とみなされる細胞加工の基本概念
「特定細胞加工物」を理解する上で欠かせないのが、「製造」とみなされる加工の概念です。法律上、細胞に「加工」を施す行為は、新たな製品を生み出す「製造」プロセスとして扱われます。これは、元の細胞とは異なる性質や機能を持たせる、あるいは細胞を増やすといった操作が加わるためです。
したがって、特定細胞加工物に該当すると判断された場合、そのプロセスは厳格な品質管理(GCTP等)の下で行われるべき「製造行為」とみなされます。単なる医療処置の一部ではなく、製品としての品質と安全性が求められる工業的な側面を持つことを認識しておきましょう。
医療機関内での調製と企業による製造の違い
実務上、医療機関の医師が自らの責任の下で院内にて行う場合は「調製」、企業等の外部施設に委託して行う場合は「製造」と呼び分けられることがあります。しかし、特定細胞加工物としての「モノの定義」自体は、場所や実施主体によって変わるものではありません。
- 医療機関内での調製: 医師の管理下で行われるが、特定細胞加工物の製造届出が必要な場合がある
- 企業による製造: 特定細胞加工物製造業の許可または認定が必要
いずれの場合も、対象となる細胞加工物が「特定細胞加工物」の定義に当てはまる限り、法が求める安全性基準を満たす必要がある点に変わりはありません。
特定細胞加工物に該当する「加工」の具体的な判断基準

「特定細胞加工物」に該当するか否かの分かれ道は、実施する操作が「加工」とみなされるかどうかにあります。厚生労働省のガイドラインや通知に基づき、どのような操作が加工に該当するのか、その具体的な判断基準を見ていきましょう。これらは細胞の生物学的特性や構造に影響を与える操作であり、再生医療の有効性と安全性に直結する重要な要素です。
人工的な増殖(培養)を伴う処理
最も代表的な「加工」の一つが、細胞の人工的な増殖、すなわち培養です。体外に取り出した細胞を、培地を用いて数や量を増やす操作は、細胞の生理的な状態を大きく変化させる可能性があるため、明確に加工とみなされます。
例えば、少量の組織から幹細胞を分離し、それを数週間かけて数千万個レベルまで増やすような工程は、典型的な特定細胞加工物の製造プロセスです。この過程では、細胞の変質や汚染のリスク管理が厳格に求められます。
細胞の活性化を目的とした薬剤処理
細胞の数を増やさない場合でも、薬剤等を用いて細胞の生理的機能を意図的に高めたり変化させたりする操作は「加工」に該当します。これを「活性化」と呼びます。
具体例としては、リンパ球を採取し、特定の抗体やサイトカインと共に培養して、がん細胞への攻撃能力を高めるような処理が挙げられます。細胞の性質そのものに人為的な介入を行っているため、元の細胞とは同等でないと判断され、規制の対象となります。
生物学的特性の改変を伴う操作
細胞の分化能や形質を変化させる操作も、特定細胞加工物の要件を満たす「加工」です。これには、未分化な細胞を特定の組織細胞へと誘導する場合や、逆に分化した細胞を初期化する場合が含まれます。
- 分化誘導: iPS細胞から神経細胞や心筋細胞を作成する
- 脱分化: 体細胞からiPS細胞を作成する
このように細胞の運命(Fate)を人為的に操作することは、高度な加工技術であり、安全性確保法上の管理が必須となります。
細胞以外の構造物との結合・形成
細胞単体だけでなく、細胞を支持体(スキャフォールド)や他の材料と組み合わせて構造化する操作も「加工」に含まれます。これは、組織工学的な手法を用いて、立体的な組織や臓器に近い構造を作り出す場合などが該当します。
例えば、培養皮膚のようにコラーゲンゲルの中に細胞を播種してシート状に形成する場合や、生分解性ポリマーに細胞を接着させて移植用グラフトを作成する場合などです。細胞と足場材料との相互作用を含めた品質評価が必要となります。
遺伝子の導入または改変操作
細胞に対して遺伝子導入を行ったり、ゲノム編集技術を用いて遺伝子を改変したりする操作は、生物学的特性を根本から変える高度な「加工」です。これはCAR-T療法のような遺伝子治療等技術にも関連します。
ウイルスベクターやプラスミドを用いて外来遺伝子を導入する操作は、細胞の機能増強や修正を目的としていますが、腫瘍化リスクなどの安全性懸念も高いため、特定細胞加工物の中でも特に慎重な取り扱いと、厳格な規制区分(第1種再生医療等)での管理が求められます。
特定細胞加工物に該当しない「最小限の操作」の範囲

一方で、細胞や組織に対するすべての操作が「加工」とみなされるわけではありません。本来の性質を変えない範囲での処理は「最小限の操作(Minimal Manipulation)」として扱われ、特定細胞加工物の定義から除外される場合があります。この境界線を正しく理解することで、過剰な申請業務を回避し、適切な法規制の下で運用することが可能になります。ここでは、加工に該当しないとされる主な操作について解説します。
組織または細胞の洗浄
採取した組織や細胞に付着している血液成分や不要な物質を生理食塩水や緩衝液などで洗い流す行為は、細胞の性質を変えるものではないため、加工には当たりません。
これは手術室や処置室で日常的に行われる操作と同様であり、清浄化を目的とした物理的な処理とみなされます。ただし、洗浄液に細胞の生理的機能を変化させるような薬剤が含まれている場合は、加工とみなされる可能性があるため注意が必要です。
組織の始末(トリミング)および裁断(ミンチ)
組織から不要な部分(脂肪や結合組織など)を取り除くトリミングや、酵素処理を容易にするためにハサミ等で細かく刻む(ミンチ)操作は、組織の構造を根本的に破壊するものでなければ、最小限の操作の範囲内とされます。
これらは、あくまで細胞を取り出しやすくするため、あるいは移植に適した形状にするための物理的な前処理であり、細胞そのものの生物学的特性を改変する意図はないと判断されるためです。
組織または細胞の凍結および解凍
細胞や組織を将来の使用のために凍結保存し、使用時に解凍する操作も、一般的には加工には該当しません。これは細胞の時間を止めるだけの操作であり、細胞の性質を変えるものではないからです。
ただし、凍結保護剤の使用は認められていますが、その過程で細胞を選別したり、特殊な薬剤で処理したりする場合は加工となる可能性があります。単なる保存・保管を目的とした操作であれば、規制対象外となります。
遠心分離による細胞の分離
血液や組織液などから、比重の違いを利用して特定の細胞層(例えば単核球層など)を回収するための遠心分離は、分離・選別の範疇であり、加工とはみなされません。
PRP(多血小板血漿)療法における遠心分離もこれに該当します。細胞そのものに手を加えているわけではなく、物理的な手段で細胞を集めているに過ぎないため、最小限の操作として扱われます。
濾過による不純物の除去
細胞懸濁液から凝集塊や組織片、異物などを取り除くためにフィルターを通す濾過操作も、加工には当たりません。これは洗浄と同様に、目的とする細胞を純化するための物理的な精製工程とみなされます。
ただし、特定の細胞表面マーカーを認識して細胞を選別・除去するような高度な分離技術を用いる場合は、その操作の意図や細胞への影響度合いによって判断が分かれることがあるため、専門的な確認が推奨されます。
混入微生物の殺菌(ガンマ線照射等)
採取した組織や細胞に対して、混入している可能性のある細菌やウイルスを不活化するために行うガンマ線照射や電子線照射などの殺菌操作も、細胞の利用目的を損なわない範囲であれば、最小限の操作に含まれます。
これは安全性を確保するための処理であり、細胞の生物学的特性を積極的に変えることを目的としていないためです。ただし、細胞自体へのダメージが大きく、細胞の機能が著しく損なわれるような強度の照射は避けなければなりません。
抗生物質の添加(輸送等の保全目的)
採取場所から加工施設への輸送時において、細菌の増殖を抑制するために抗生物質を添加することは、適切な保存措置とは認められません。
食品衛生法に基づく「食品、添加物等の規格基準」(厚生労働省告示第370号)では、抗生物質や化学的合成品である抗菌性物質の含有は原則として禁止されており、これは輸送や保存を目的とする場合でも例外ではないからです。
細胞の品質維持を意図した措置であっても、法令に抵触するリスクがあるため、安易な添加は避けるべきでしょう。
輸送中の保全については、薬剤の使用ではなく、冷蔵や冷凍といった物理的な温度管理によって品質を維持することが推奨されます。
特定細胞加工物の定義や関連する規制を正確に把握し、コンプライアンスを遵守した運用を心がけてみてください。
リスク区分による特定細胞加工物の分類(第1種・第2種・第3種)

特定細胞加工物は、その細胞が持つリスクの程度に応じて、第1種から第3種までの3つのカテゴリーに分類されます。この区分によって、再生医療等提供計画の審査を行う委員会の種類や、厚生労働大臣への届出・申請の手続きが異なります。自社が取り扱う特定細胞加工物がどのリスク区分に該当するかを正しく理解することは、適切な法的手続きを進める上で不可欠です。
第1種:ヒトES細胞やiPS細胞、他家細胞等(高リスク)
第1種は、これまでヒトに実施された実績が極めて少ない、あるいは高いリスクが想定される技術が該当します。これには、ES細胞(胚性幹細胞)やiPS細胞(人工多能性幹細胞)のような多能性幹細胞を用いる場合や、他人の細胞(他家細胞)を用いる場合が含まれます。
また、動物の細胞を利用する場合や、遺伝子導入を行った細胞もこの区分に入ります。第1種に該当する場合は、特定認定再生医療等委員会での審査を経て、厚生労働大臣への提供計画の提出が必要となり、最も厳格な手続きが求められます。
第2種:体性幹細胞等(中リスク)
第2種は、中程度のリスクがあるとされる技術で、主に患者自身の体性幹細胞(脂肪由来幹細胞など)を培養して用いる場合が該当します。現在、美容医療や整形外科領域で広く行われている再生医療の多くがこの区分に含まれます。
培養によって細胞の性質が変化する可能性があるため、安全性と有効性の慎重な確認が必要です。第2種の手続きでは、特定認定再生医療等委員会での審査が必要となりますが、第1種と比較すると、既にある程度の実績がある技術が含まれています。
第3種:体細胞加工等(低リスク)
第3種は、細胞の加工を行うものの、リスクが比較的低いとされる技術です。例えば、がん免疫療法のように、患者自身の体細胞(幹細胞ではない細胞)を培養・加工して用いる場合などがこれに当たります。
元の細胞が分化能を持たない、あるいは限定的であるため、腫瘍化などのリスクが低いと考えられています。第3種の場合は、認定再生医療等委員会での審査で足りますが、特定細胞加工物としての製造管理・品質管理の基準は遵守しなければなりません。
特定細胞加工物に該当する場合に事業者が講ずべき措置

取り扱う細胞が「特定細胞加工物」に該当すると判断された場合、事業者は再生医療安全性確保法に基づき、厳格な措置を講じる義務が生じます。これは患者様の安全を守るための必須条件であり、違反した場合は事業の停止命令や罰則の対象となります。ここでは、特定細胞加工物を製造・提供する事業者が必ずクリアしなければならない、主要な法的要件について解説します。
特定細胞加工物製造許可の取得(法第35条)
特定細胞加工物を製造しようとする事業者は、国内の製造施設ごとに、厚生労働大臣からの「特定細胞加工物製造許可」を取得しなければなりません(法第35条)。
この許可を取得するためには、申請書に構造設備の概要や製造管理の方法などを記載して提出し、実地調査等を受ける必要があります。許可は5年ごとの更新制となっており、継続的な基準適合が求められます。なお、医療機関内で医師が自らの患者のために調製する場合であっても、届出が必要となるケースがあるため確認が必要です。
海外における特定細胞加工物製造施設の認定(法第39条)
海外の施設で製造された特定細胞加工物を輸入して用いる場合、その海外製造施設は厚生労働大臣の「認定」を受ける必要があります(法第39条)。これは国内の製造許可に相当するもので、海外施設であっても日本の法律が求める水準の安全性や品質管理体制が整っていることを担保するためです。
海外CDMO(医薬品開発製造受託機関)などに製造委託を検討している場合は、その施設が日本の認定を取得しているか、あるいは取得可能かを確認することが事業計画上の重要なマイルストーンとなります。
構造設備基準(GCTP省令)への適合
製造施設は、「特定細胞加工物製造業者の構造設備の基準(GCTP省令)」に適合していなければなりません。これには、無菌操作を行うためのクリーンルームの設置、空調システムの管理、作業室とその他の区域の明確な区分けなどが含まれます。
ハード面での要件は非常に細かく規定されており、交叉汚染(クロスコンタミネーション)や異物混入を確実に防止できる設計であることが求められます。既存の施設を改修する場合でも、この基準を満たすための設備投資が必要となるでしょう。
製造管理および品質管理の基準遵守
設備だけでなく、運用面(ソフト面)においても「製造管理及び品質管理の基準」を遵守することが義務付けられています。これには、標準操作手順書(SOP)の作成、製造記録・品質試験記録の保管、衛生管理、苦情処理などの体制整備が含まれます。
また、製造管理者などの責任者を配置し、組織として品質を保証するシステム(QMS)を構築しなければなりません。特定細胞加工物は製品ごとのばらつきが出やすいため、プロセス全体を通じた厳密な管理が製品の信頼性を支えることになります。
まとめ

本記事では、再生医療安全性確保法における「特定細胞加工物の定義」について、法的根拠から具体的な判断基準、リスク区分、そして事業者が講ずべき措置までを解説いたしました。
重要なポイントは以下の通りです。
- 定義の核心: 人為的な「加工(培養、活性化、形質転換等)」が施された細胞であること。
- 境界線: 洗浄、凍結、遠心分離などの「最小限の操作」は加工に含まれない。
- 事業者の義務: 該当する場合は、製造許可の取得やGCTP準拠の構造設備・管理体制が必須となる。
特定細胞加工物の該当性は、個別の事例によって判断が難しいグレーゾーンも存在します。自己判断のみで進めることなく、PMDA(医薬品医療機器総合機構)の事前面談や、再生医療に精通した専門家への相談を活用し、確実なコンプライアンス体制を築くことを強くお勧めいたします。正確な法的理解こそが、安全で信頼される再生医療事業の礎となるでしょう。
特定細胞加工物の定義についてよくある質問

特定細胞加工物の定義や取り扱いに関して、再生医療の現場や薬事担当者の方から頻繁に寄せられる質問をまとめました。実務における判断の参考としてご活用ください。
よくある質問
-
Q1. PRP(多血小板血漿)療法で用いる加工物は、特定細胞加工物に該当しますか?
- 一般的に、PRP療法で用いられるPRPは、血液の遠心分離(最小限の操作)によって得られるものであり、薬剤による活性化や培養を行わない限り、特定細胞加工物には該当しません。ただし、第3種再生医療等としての届出は必要です。
-
Q2. 医療機関内で医師が自ら培養を行う場合でも、製造業の許可は必要ですか?
- 医療機関内で医師が自らの患者のために培養(調製)を行う場合は、製造業の「許可」ではなく、厚生労働大臣への「届出」が必要となります。ただし、他の医療機関からの委託を受けて培養する場合は「許可」が必要です。
-
Q3. 組織から細胞を分離するために酵素処理を行うことは「加工」になりますか?
- 組織構造を分解して細胞を取り出すための酵素処理は、一般的に細胞の生物学的特性を変えない範囲であれば「最小限の操作」とみなされることが多いですが、処理の強度や目的によっては加工と判断される場合もあるため、慎重な確認が必要です。
-
Q4. 細胞の輸送中に温度管理を行うことは「加工」に含まれますか?
- いいえ、含まれません。輸送中の温度管理は細胞の品質を維持・保存するための措置であり、細胞の性質を改変する操作ではないため、加工には該当しません。
-
Q5. 海外から特定細胞加工物を輸入する場合、どのような手続きが必要ですか?
- 輸入する特定細胞加工物の製造元である海外施設が、日本の厚生労働大臣から「認定(海外特定細胞加工物製造事業者認定)」を受けている必要があります。また、輸入者は国内での提供計画にその旨を記載する必要があります。